CELLxGENE: scRNA-seq¶
CZ CELLxGENE hosts the globally largest standardized collection of scRNA-seq datasets.
LaminDB makes it easy to query the CELLxGENE data and integrate it with in-house data of any kind (omics, phenotypes, pdfs, notebooks, ML models, …).
You can use the CELLxGENE data in two ways:
Query collections of
AnnDataobjects.Slice a big array store produced by concatenated
AnnDataobjects viatiledbsoma.
If you are interested in building similar data assets in-house:
See the transfer guide to zero-copy data to your own LaminDB instance.
See the scRNA guide to create a growing, standardized & versioned scRNA-seq dataset collection.
Show me a screenshot
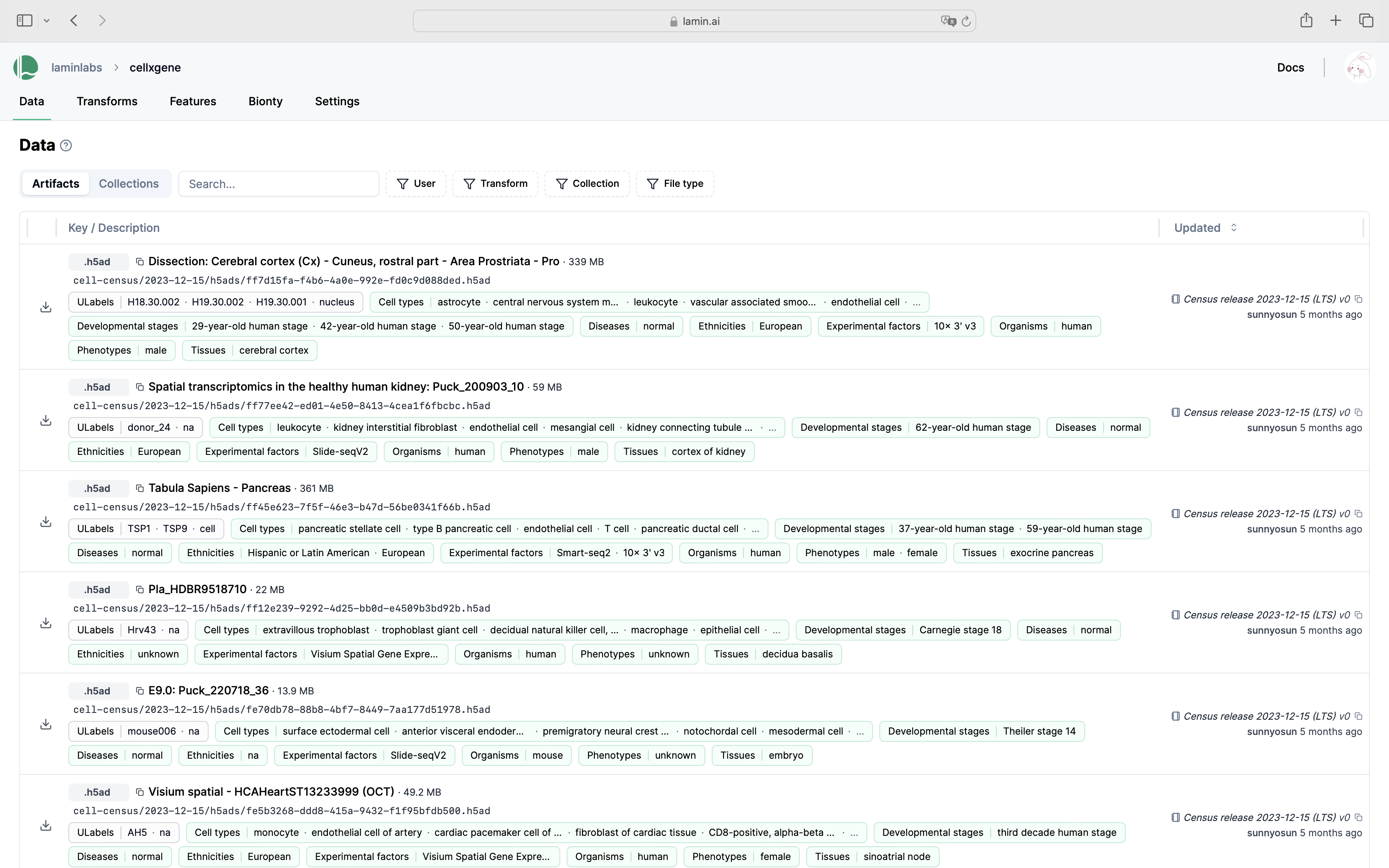
Load the public LaminDB instance that mirrors cellxgene:
# !pip install 'lamindb[bionty,ourprojects,jupyter]'
!lamin load laminlabs/cellxgene
Show code cell output
! schema module 'ourprojects' is not installed → no access to its labels & registries (resolve via `pip install ourprojects`)
→ connected lamindb: laminlabs/cellxgene
import lamindb as ln
import bionty as bt
Show code cell output
! schema module 'ourprojects' is not installed → no access to its labels & registries (resolve via `pip install ourprojects`)
→ connected lamindb: laminlabs/cellxgene
Query & understand metadata¶
Auto-complete metadata¶
You can create look-up objects for any registry in LaminDB, including basic biological entities and things like users or storage locations.
Let’s use auto-complete to look up cell types:
Show me a screenshot
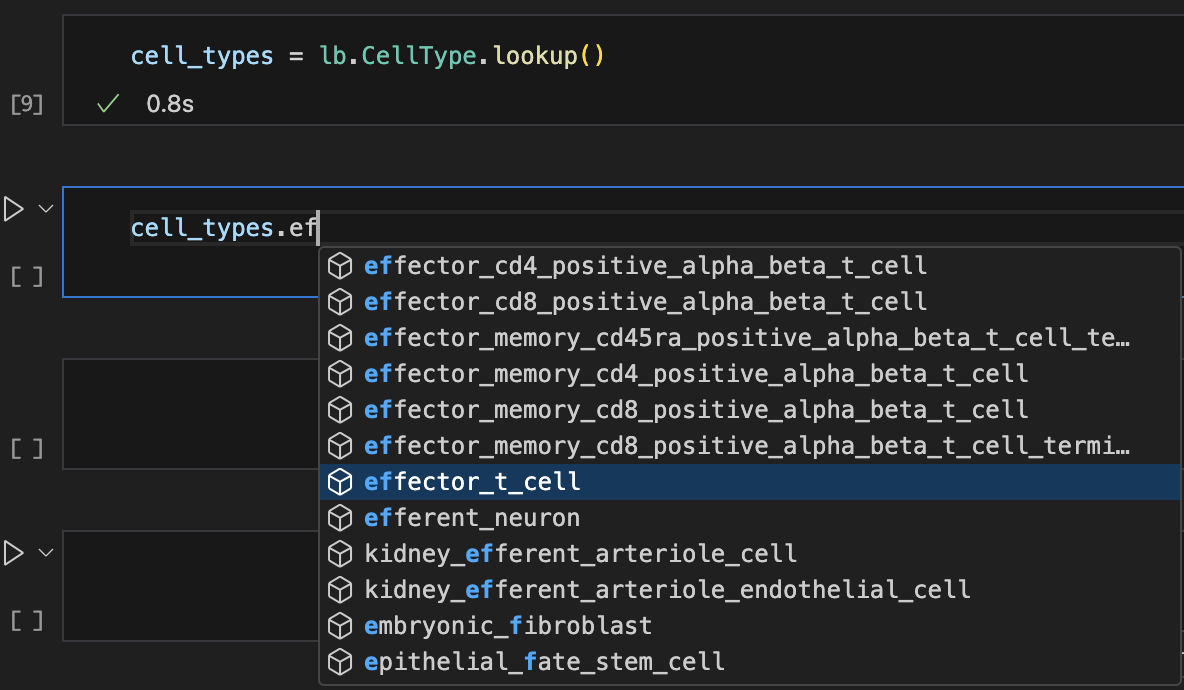
cell_types = bt.CellType.lookup()
cell_types.effector_t_cell
Show code cell output
CellType(uid='3nfZTVV4', name='effector T cell', ontology_id='CL:0000911', synonyms='effector T-cell|effector T-lymphocyte|effector T lymphocyte', description='A Differentiated T Cell With Ability To Traffic To Peripheral Tissues And Is Capable Of Mounting A Specific Immune Response.', created_by_id=1, source_id=48, created_at=2023-11-28 22:30:57 UTC)
You can also arbitrarily chain filters and create lookups from them:
users = ln.User.lookup()
organisms = bt.Organism.lookup()
experimental_factors = bt.ExperimentalFactor.lookup() # labels for experimental factors
tissues = bt.Tissue.lookup() # tissue labels
suspension_types = ln.ULabel.filter(name="is_suspension_type").one().children.lookup() # suspension types
# here we choose to return .name directly
features = ln.Feature.lookup(return_field="name")
assays = bt.ExperimentalFactor.lookup(return_field="name")
Search & filter metadata¶
We can use search & filters for metadata:
bt.CellType.search("effector T cell").df().head()
Show code cell output
| uid | name | ontology_id | abbr | synonyms | description | source_id | run_id | created_at | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|
| id | ||||||||||
| 1623 | 3nfZTVV4 | effector T cell | CL:0000911 | None | effector T-cell|effector T-lymphocyte|effector... | A Differentiated T Cell With Ability To Traffi... | 48 | None | 2023-11-28 22:30:57.481760+00:00 | 1 |
| 1169 | 6JD5JCZC | CD8-positive, alpha-beta cytokine secreting ef... | CL:0000908 | None | CD8-positive, alpha-beta cytokine secreting ef... | A Cd8-Positive, Alpha-Beta T Cell With The Phe... | 48 | None | 2023-11-28 22:27:55.571572+00:00 | 1 |
| 1229 | 69TEBGqb | exhausted T cell | CL:0011025 | None | Tex cell|An effector T cell that displays impa... | None | 48 | None | 2023-11-28 22:27:55.572880+00:00 | 1 |
| 1331 | 43cBCa7s | helper T cell | CL:0000912 | None | helper T-lymphocyte|T-helper cell|helper T lym... | A Effector T Cell That Provides Help In The Fo... | 48 | None | 2023-11-28 22:27:55.575949+00:00 | 1 |
| 1503 | 1oa5G2Mq | memory T cell | CL:0000813 | None | memory T-cell|memory T lymphocyte|memory T-lym... | A Long-Lived, Antigen-Experienced T Cell That ... | 48 | None | 2023-11-28 22:27:55.580286+00:00 | 1 |
And use a uid to filter exactly one metadata record:
effector_t_cell = bt.CellType.get("3nfZTVV4")
effector_t_cell
Show code cell output
CellType(uid='3nfZTVV4', name='effector T cell', ontology_id='CL:0000911', synonyms='effector T-cell|effector T-lymphocyte|effector T lymphocyte', description='A Differentiated T Cell With Ability To Traffic To Peripheral Tissues And Is Capable Of Mounting A Specific Immune Response.', created_by_id=1, source_id=48, created_at=2023-11-28 22:30:57 UTC)
Understand ontologies¶
View the related ontology terms:
effector_t_cell.view_parents(distance=2, with_children=True)
Show code cell output

Or access them programmatically:
effector_t_cell.children.df()
Show code cell output
| uid | name | ontology_id | abbr | synonyms | description | source_id | run_id | created_at | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|
| id | ||||||||||
| 931 | 2VQirdSp | effector CD8-positive, alpha-beta T cell | CL:0001050 | None | effector CD8-positive, alpha-beta T lymphocyte... | A Cd8-Positive, Alpha-Beta T Cell With The Phe... | 48 | None | 2023-11-28 22:27:55.565976+00:00 | 1 |
| 1088 | 490Xhb24 | effector CD4-positive, alpha-beta T cell | CL:0001044 | None | effector CD4-positive, alpha-beta T lymphocyte... | A Cd4-Positive, Alpha-Beta T Cell With The Phe... | 48 | None | 2023-11-28 22:27:55.569828+00:00 | 1 |
| 1229 | 69TEBGqb | exhausted T cell | CL:0011025 | None | Tex cell|An effector T cell that displays impa... | None | 48 | None | 2023-11-28 22:27:55.572880+00:00 | 1 |
| 1309 | 5s4gCMdn | cytotoxic T cell | CL:0000910 | None | cytotoxic T lymphocyte|cytotoxic T-lymphocyte|... | A Mature T Cell That Differentiated And Acquir... | 48 | None | 2023-11-28 22:27:55.575440+00:00 | 1 |
| 1331 | 43cBCa7s | helper T cell | CL:0000912 | None | helper T-lymphocyte|T-helper cell|helper T lym... | A Effector T Cell That Provides Help In The Fo... | 48 | None | 2023-11-28 22:27:55.575949+00:00 | 1 |
Query for individual datasets¶
Every individual dataset in CELLxGENE is an .h5ad file that is stored as an artifact in LaminDB. Here is an exemplary query:
ln.Artifact.filter(
suffix=".h5ad", # filename suffix
description__contains="immune",
size__gt=1e9, # size > 1GB
cell_types__in=[cell_types.b_cell, cell_types.t_cell], # cell types measured in AnnData
created_by=users.sunnyosun # creator
).order_by(
"created_at"
).df(
include=["cell_types__name", "created_by__handle"] # join with additional info
).head()
Show code cell output
| cell_types__name | created_by__handle | uid | version | is_latest | description | key | suffix | type | size | ... | n_observations | _hash_type | _accessor | visibility | _key_is_virtual | storage_id | transform_id | run_id | created_at | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 879 | [conventional dendritic cell, classical monocy... | sunnyosun | BCutg5cxmqLmy2Z5SS8J | 2023-07-25 | False | Type I interferon autoantibodies are associate... | cell-census/2023-07-25/h5ads/01ad3cd7-3929-465... | .h5ad | None | 6353682597 | ... | 600929 | md5-n | AnnData | 1 | False | 2 | 11 | 16 | 2023-11-28 21:45:49.000689+00:00 | 1 |
| 1106 | [immature B cell, monocyte, naive thymus-deriv... | sunnyosun | 3xdOASXuAxxJtSchJO3D | 2023-07-25 | False | HSC/immune cells (all hematopoietic-derived ce... | cell-census/2023-07-25/h5ads/48101fa2-1a63-451... | .h5ad | None | 6214230662 | ... | 589390 | md5-n | AnnData | 1 | False | 2 | 11 | 16 | 2023-11-28 21:46:02.770448+00:00 | 1 |
| 1174 | [monocyte, conventional dendritic cell, plasma... | sunnyosun | wt7eD72sTzwL3rfYaZr2 | 2023-07-25 | False | A scRNA-seq atlas of immune cells at the CNS b... | cell-census/2023-07-25/h5ads/58b01044-c5e5-4b0... | .h5ad | None | 1052158249 | ... | 130908 | md5-n | AnnData | 1 | False | 2 | 11 | 16 | 2023-11-28 21:46:06.866895+00:00 | 1 |
| 1377 | [monocyte, ciliated cell, macrophage, natural ... | sunnyosun | znTBqWgfYgFlLjdQ6Ba7 | 2023-07-25 | False | Large-scale single-cell analysis reveals criti... | cell-census/2023-07-25/h5ads/9dbab10c-118d-496... | .h5ad | None | 13929140098 | ... | 1462702 | md5-n | AnnData | 1 | False | 2 | 11 | 16 | 2023-11-28 21:46:19.141781+00:00 | 1 |
| 1482 | [effector CD4-positive, alpha-beta T cell, con... | sunnyosun | dEP0dZ8UxLgwnkLjz6Iq | 2023-07-25 | False | Single-cell sequencing links multiregional imm... | cell-census/2023-07-25/h5ads/bd65a70f-b274-413... | .h5ad | None | 1204103287 | ... | 167283 | md5-n | AnnData | 1 | False | 2 | 11 | 16 | 2023-11-28 21:46:25.468183+00:00 | 1 |
5 rows × 22 columns
What happens under the hood?
As you saw from inspecting ln.Artifact, ln.Artifact.cell_types relates artifacts with bt.CellType.
The expression cell_types__name__in performs the join of the underlying registries and matches bt.CellType.name to ["B cell", "T cell"].
Similar for created_by, which relates artifacts with ln.User.
To see what you can query for, look at the registry representation.
ln.Artifact
Show code cell output
Artifact
Simple fields
.uid: CharField
.description: CharField
.key: CharField
.suffix: CharField
.type: CharField
.size: BigIntegerField
.hash: CharField
.n_objects: BigIntegerField
.n_observations: BigIntegerField
.visibility: SmallIntegerField
.version: CharField
.is_latest: BooleanField
.created_at: DateTimeField
.updated_at: DateTimeField
Relational fields
.storage: Storage
.transform: Transform
.run: Run
.created_by: User
.ulabels: ULabel
.input_of_runs: Run
.feature_sets: FeatureSet
.collections: Collection
Bionty fields
.organisms: bionty.Organism
.genes: bionty.Gene
.proteins: bionty.Protein
.cell_markers: bionty.CellMarker
.tissues: bionty.Tissue
.cell_types: bionty.CellType
.diseases: bionty.Disease
.cell_lines: bionty.CellLine
.phenotypes: bionty.Phenotype
.pathways: bionty.Pathway
.experimental_factors: bionty.ExperimentalFactor
.developmental_stages: bionty.DevelopmentalStage
.ethnicities: bionty.Ethnicity
Slice an individual dataset¶
Let’s look at an artifact and show its metadata using .describe().
artifact = ln.Artifact.get(description="Mature kidney dataset: immune", is_latest=True)
artifact.describe()
Show code cell output
Artifact(uid='WwmBIhBNLTlRcSoBDt76', version='2024-07-01', is_latest=True, description='Mature kidney dataset: immune', key='cell-census/2024-07-01/h5ads/20d87640-4be8-487f-93d4-dce38378d00f.h5ad', suffix='.h5ad', type='dataset', size=45158726, hash='GCMHkdQSTeXxRVF7gMZFIA', n_observations=7803, _hash_type='md5-n', _accessor='AnnData', visibility=1, _key_is_virtual=False, created_at=2024-07-12 12:34:09 UTC)
Provenance
.storage = 's3://cellxgene-data-public'
.transform = 'Census release 2024-07-01 (LTS)'
.run = '2024-07-16 12:49:41 UTC'
.created_by = 'sunnyosun'
Labels
.organisms = 'human'
.tissues = 'kidney blood vessel', 'renal pelvis', 'cortex of kidney', 'renal medulla', 'kidney'
.cell_types = 'CD8-positive, alpha-beta T cell', 'mature NK T cell', 'CD4-positive, alpha-beta T cell', 'natural killer cell', 'non-classical monocyte', 'plasmacytoid dendritic cell', 'neutrophil', 'B cell', 'kidney resident macrophage', 'dendritic cell', ...
.diseases = 'normal'
.phenotypes = 'male', 'female'
.experimental_factors = '10x 3' v2'
.developmental_stages = '2-year-old human stage', '4-year-old human stage', '12-year-old human stage', '44-year-old human stage', '49-year-old human stage', '53-year-old human stage', '63-year-old human stage', '64-year-old human stage', '67-year-old human stage', '70-year-old human stage', ...
.ethnicities = 'unknown'
.ulabels = 'TxK2', 'Wilms1', 'TxK4', 'TTx', 'RCC3', 'RCC1', 'VHL', 'TxK3', 'TxK1', 'Wilms3', ...
Feature sets
'obs' = 'assay', 'cell_type', 'development_stage', 'disease', 'donor_id', 'self_reported_ethnicity', 'sex', 'suspension_type', 'tissue', 'organism', 'tissue_type'
'var' = 'None', 'EBF1', 'LINC02202', 'RNF145', 'LINC01932', 'UBLCP1', 'IL12B', 'LINC01845', 'LINC01847', 'ADRA1B', 'TTC1', 'PWWP2A', 'FABP6', 'FABP6-AS1', 'CCNJL', 'C1QTNF2'
Feature values -- internal
'assay' = 10x 3' v2
'cell_type' = B cell, CD4-positive, alpha-beta T cell, CD8-positive, alpha-beta T cell, classical monocyte, dendritic cell, kidney resident macrophage, mast cell, mature NK T cell, natural killer cell, neutrophil, ...
'development_stage' = 12-year-old human stage, 19-month-old human stage, 2-year-old human stage, 4-year-old human stage, 44-year-old human stage, 49-year-old human stage, 53-year-old human stage, 63-year-old human stage, 64-year-old human stage, 67-year-old human stage, ...
'disease' = normal
'donor_id' = RCC1, RCC2, RCC3, TTx, TxK1, TxK2, TxK3, TxK4, VHL, Wilms1, ...
'organism' = human
'self_reported_ethnicity' = unknown
'sex' = female, male
'suspension_type' = cell
'tissue' = cortex of kidney, kidney, kidney blood vessel, renal medulla, renal pelvis
More ways of accessing metadata
Access just features:
artifact.features
Or get labels given a feature:
artifact.labels.get(features.tissue).df()
If you want to query a slice of the array data, you have two options:
Cache the artifact on disk and return the path to the cached data. Doesn’t download anything if the artifact is already in the cache.
Cache & load the entire artifact into memory via
artifact.load() -> AnnDataStream the array using a (cloud-backed) accessor
artifact.open() -> AnnDataAccessor
Both will run much faster in the AWS us-west-2 data center.
Cache:
cache_path = artifact.cache()
cache_path
Show code cell output
! run input wasn't tracked, call `ln.track()` and re-run
PosixUPath('/home/runner/.cache/lamindb/cellxgene-data-public/cell-census/2024-07-01/h5ads/20d87640-4be8-487f-93d4-dce38378d00f.h5ad')
Cache & load:
adata = artifact.load()
adata
Show code cell output
! run input wasn't tracked, call `ln.track()` and re-run
AnnData object with n_obs × n_vars = 7803 × 32839
obs: 'donor_id', 'donor_age', 'self_reported_ethnicity_ontology_term_id', 'organism_ontology_term_id', 'sample_uuid', 'tissue_ontology_term_id', 'development_stage_ontology_term_id', 'suspension_uuid', 'suspension_type', 'library_uuid', 'assay_ontology_term_id', 'mapped_reference_annotation', 'is_primary_data', 'cell_type_ontology_term_id', 'author_cell_type', 'disease_ontology_term_id', 'reported_diseases', 'sex_ontology_term_id', 'compartment', 'Experiment', 'Project', 'tissue_type', 'cell_type', 'assay', 'disease', 'organism', 'sex', 'tissue', 'self_reported_ethnicity', 'development_stage', 'observation_joinid'
var: 'feature_is_filtered', 'feature_name', 'feature_reference', 'feature_biotype', 'feature_length'
uns: 'citation', 'default_embedding', 'schema_reference', 'schema_version', 'title'
obsm: 'X_umap'
Now we have an AnnData object, which stores observation annotations matching our artifact-level query in the .obs slot, and we can re-use almost the same query on the array-level.
See the array-level query
adata_slice = adata[
adata.obs.cell_type.isin(
[cell_types.dendritic_cell.name, cell_types.neutrophil.name]
)
& (adata.obs.tissue == tissues.kidney.name)
& (adata.obs.suspension_type == suspension_types.cell.name)
& (adata.obs.assay == experimental_factors.ln_10x_3_v2.name)
]
adata_slice
See the artifact-level query
collection = ln.Collection.filter(name="cellxgene-census", version="2024-07-01").one()
query = collection.artifacts.filter(
organism=organisms.human,
cell_types__in=[cell_types.dendritic_cell, cell_types.neutrophil],
tissues=tissues.kidney,
ulabels=suspension_types.cell,
experimental_factors=experimental_factors.ln_10x_3_v2,
)
AnnData uses pandas to manage metadata and the syntax differs slightly. However, the same metadata records are used.
Stream, slice and load the slice into memory:
with artifact.open() as adata_backed:
display(adata_backed)
Show code cell output
! run input wasn't tracked, call `ln.track()` and re-run
AnnDataAccessor object with n_obs × n_vars = 7803 × 32839
constructed for the AnnData object 20d87640-4be8-487f-93d4-dce38378d00f.h5ad
obs: ['Experiment', 'Project', '_index', 'assay', 'assay_ontology_term_id', 'author_cell_type', 'cell_type', 'cell_type_ontology_term_id', 'compartment', 'development_stage', 'development_stage_ontology_term_id', 'disease', 'disease_ontology_term_id', 'donor_age', 'donor_id', 'is_primary_data', 'library_uuid', 'mapped_reference_annotation', 'observation_joinid', 'organism', 'organism_ontology_term_id', 'reported_diseases', 'sample_uuid', 'self_reported_ethnicity', 'self_reported_ethnicity_ontology_term_id', 'sex', 'sex_ontology_term_id', 'suspension_type', 'suspension_uuid', 'tissue', 'tissue_ontology_term_id', 'tissue_type']
obsm: ['X_umap']
raw: ['X', 'var', 'varm']
uns: ['citation', 'default_embedding', 'schema_reference', 'schema_version', 'title']
var: ['_index', 'feature_biotype', 'feature_is_filtered', 'feature_length', 'feature_name', 'feature_reference']
We now have an AnnDataAccessor object, which behaves much like an AnnData, and slicing looks similar to the query above.
See the slicing operation
adata_backed_slice = adata_backed[
adata_backed.obs.cell_type.isin(
[cell_types.dendritic_cell.name, cell_types.neutrophil.name]
)
& (adata_backed.obs.tissue == tissues.kidney.name)
& (adata_backed.obs.suspension_type == suspension_types.cell.name)
& (adata_backed.obs.assay == experimental_factors.ln_10x_3_v2.name)
]
adata_backed_slice.to_memory()
Query collections of datasets¶
Let’s search collections from CELLxGENE within the 2024-07-01 release:
ln.Collection.filter(version="2024-07-01").search("human retina", limit=10)
Show code cell output
<QuerySet [Collection(uid='2gBKIwx8AtCHc4nfcQqc', version='2024-07-01', is_latest=True, name='A single-cell transcriptome atlas of the adult human retina', description='10.15252/embj.2018100811', hash='sCh4gUTJJJjECsp1dj0q', reference='3472f32d-4a33-48e2-aad5-666d4631bf4c', reference_type='CELLxGENE Collection ID', visibility=1, created_by_id=1, transform_id=22, run_id=27, created_at=2024-07-16 12:20:47 UTC), Collection(uid='zZLyhpo1aDdxdbULFbVT', version='2024-07-01', is_latest=True, name='Single-cell transcriptomic atlas of the human retina identifies cell types associated with age-related macular degeneration', description='10.1038/s41467-019-12780-8', hash='1B0m9_FahAvefSTM8_AV', reference='1a486c4c-c115-4721-8c9f-f9f096e10857', reference_type='CELLxGENE Collection ID', visibility=1, created_by_id=1, transform_id=22, run_id=27, created_at=2024-07-16 12:20:25 UTC), Collection(uid='tZYmzwfh0bIYzKBQVuro', version='2024-07-01', is_latest=True, name='Cell Types of the Human Retina and Its Organoids at Single-Cell Resolution', description='10.1016/j.cell.2020.08.013', hash='nGcCV4HJONcma2SExXw2', reference='2f4c738f-e2f3-4553-9db2-0582a38ea4dc', reference_type='CELLxGENE Collection ID', visibility=1, created_by_id=1, transform_id=22, run_id=27, created_at=2024-07-16 12:19:55 UTC), Collection(uid='8ohRJQq8e3F7pdlBZbhz', version='2024-07-01', is_latest=True, name='Single cell atlas of the human retina', description='10.1101/2023.11.07.566105', hash='_vU7tll3t-0NCuJL-fm0', reference='4c6eaf5c-6d57-4c76-b1e9-60df8c655f1e', reference_type='CELLxGENE Collection ID', visibility=1, created_by_id=1, transform_id=22, run_id=27, created_at=2024-07-16 12:19:25 UTC), Collection(uid='quQDnLsMLkP3JRsC8gp4', version='2024-07-01', is_latest=True, name='Single-cell transcriptomic atlas for adult human retina', description='10.1016/j.xgen.2023.100298', hash='NIo8G6_reJTEqMzW2nMc', reference='af893e86-8e9f-41f1-a474-ef05359b1fb7', reference_type='CELLxGENE Collection ID', visibility=1, created_by_id=1, transform_id=22, run_id=27, created_at=2024-07-16 12:21:51 UTC), Collection(uid='Yxth0JJgMb2VVOCfSgWj', version='2024-07-01', is_latest=True, name='Single-cell transcriptomics of the human retinal pigment epithelium and choroid in health and macular degeneration', description='10.1073/pnas.1914143116', hash='j2LqihaaNawOtEFysl3c', reference='f8057c47-fcd8-4fcf-88b0-e2f930080f6e', reference_type='CELLxGENE Collection ID', visibility=1, created_by_id=1, transform_id=22, run_id=27, created_at=2024-07-16 12:21:55 UTC)]>
Let’s get the record of the top hit collection:
collection = ln.Collection.get("quQDnLsMLkP3JRsC8gp4")
collection
Show code cell output
Collection(uid='quQDnLsMLkP3JRsC8gp4', version='2024-07-01', is_latest=True, name='Single-cell transcriptomic atlas for adult human retina', description='10.1016/j.xgen.2023.100298', hash='NIo8G6_reJTEqMzW2nMc', reference='af893e86-8e9f-41f1-a474-ef05359b1fb7', reference_type='CELLxGENE Collection ID', visibility=1, created_by_id=1, transform_id=22, run_id=27, created_at=2024-07-16 12:21:51 UTC)
It’s a Science paper and we can find more information on it using the DOI or CELLxGENE collection id. There are multiple versions of this collection.
collection.versions.df()
Show code cell output
| uid | version | is_latest | name | description | hash | reference | reference_type | visibility | transform_id | meta_artifact_id | run_id | created_at | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | ||||||||||||||
| 134 | quQDnLsMLkP3JRsC6WWz | 2023-07-25 | False | Single-cell transcriptomic atlas for adult hum... | 10.1016/j.xgen.2023.100298 | xhfSShX8lypXPx00zevx | af893e86-8e9f-41f1-a474-ef05359b1fb7 | CELLxGENE Collection ID | 1 | NaN | None | NaN | 2024-01-08 12:22:12.891930+00:00 | 1 |
| 291 | quQDnLsMLkP3JRsCJNGB | 2023-12-15 | False | Single-cell transcriptomic atlas for adult hum... | 10.1016/j.xgen.2023.100298 | FsD52kpR7dF2h78-P3ka | af893e86-8e9f-41f1-a474-ef05359b1fb7 | CELLxGENE Collection ID | 1 | 17.0 | None | 22.0 | 2024-01-11 13:41:01.880382+00:00 | 1 |
| 606 | quQDnLsMLkP3JRsC8gp4 | 2024-07-01 | True | Single-cell transcriptomic atlas for adult hum... | 10.1016/j.xgen.2023.100298 | NIo8G6_reJTEqMzW2nMc | af893e86-8e9f-41f1-a474-ef05359b1fb7 | CELLxGENE Collection ID | 1 | 22.0 | None | 27.0 | 2024-07-16 12:21:51.449109+00:00 | 1 |
The collection groups artifacts.
collection.artifacts.df()
Show code cell output
! no run & transform got linked, call `ln.track()` & re-run
! run input wasn't tracked, call `ln.track()` and re-run
| uid | version | is_latest | description | key | suffix | type | size | hash | n_objects | n_observations | _hash_type | _accessor | visibility | _key_is_virtual | storage_id | transform_id | run_id | created_at | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | ||||||||||||||||||||
| 2852 | Oc6ANFJ0FgOW1B70mNIq | 2024-07-01 | True | Photoreceptor cells in human retina (rod cells... | cell-census/2024-07-01/h5ads/00e5dedd-b9b7-43b... | .h5ad | dataset | 990594324 | qFT65q6_k30pki8-1_2HoQ | None | 21422 | md5-n | AnnData | 1 | False | 2 | 22 | 27 | 2024-07-12 12:34:08.813762+00:00 | 1 |
| 2855 | wYiUe9hn4TJijpoX90Mr | 2024-07-01 | True | All major cell types in adult human retina | cell-census/2024-07-01/h5ads/0129dbd9-a7d3-4f6... | .h5ad | dataset | 14638089351 | bXxaz_quQ4mIbVlarLZZKQ | None | 244474 | md5-n | AnnData | 1 | False | 2 | 22 | 27 | 2024-07-12 12:34:08.826175+00:00 | 1 |
| 2919 | GA2BXWwoJlcRfzNp3iyQ | 2024-07-01 | True | Horizontal cells in human retina | cell-census/2024-07-01/h5ads/11ef37ee-2173-458... | .h5ad | dataset | 404987285 | fR0O7fSUHxmAfEDC8J7Ipw | None | 7348 | md5-n | AnnData | 1 | False | 2 | 22 | 27 | 2024-07-12 12:34:08.949267+00:00 | 1 |
| 3018 | QpuY5RsGTBBMN61QGY4t | 2024-07-01 | True | Amacrine cells in human retina | cell-census/2024-07-01/h5ads/359f7af4-87d4-411... | .h5ad | dataset | 3382221253 | S7gXlC-cJ362BOqYZFxMOA | None | 56507 | md5-n | AnnData | 1 | False | 2 | 22 | 27 | 2024-07-12 12:34:09.160201+00:00 | 1 |
| 3273 | 1OyQQLNfu1nzvVADODND | 2024-07-01 | True | Bipolar cells in human retina | cell-census/2024-07-01/h5ads/8f10185b-e0b3-46a... | .h5ad | dataset | 3075818557 | 1GQwZcymSrr7d2Xit-5Deg | None | 53040 | md5-n | AnnData | 1 | False | 2 | 22 | 27 | 2024-07-12 12:34:09.644258+00:00 | 1 |
| 3378 | Ce4Mqe4X2vUhwkwnh5YQ | 2024-07-01 | True | Retinal ganglion cells in human retina | cell-census/2024-07-01/h5ads/aad97cb5-f375-45e... | .h5ad | dataset | 784580498 | w-_LJDfBv7vsZqw-9Jt72g | None | 11617 | md5-n | AnnData | 1 | False | 2 | 22 | 27 | 2024-07-12 12:34:09.816906+00:00 | 1 |
| 3600 | 80xlsVmayPPBCCEZ7aBc | 2024-07-01 | True | Non-neuronal cells in human retina | cell-census/2024-07-01/h5ads/ed419b4e-db9b-40f... | .h5ad | dataset | 1070671504 | slN6j-9aSrYFw-IPL-wv-A | None | 18011 | md5-n | AnnData | 1 | False | 2 | 22 | 27 | 2024-07-12 12:34:10.255394+00:00 | 1 |
Let’s now look at the collection that corresponds to the cellxgene-census release of .h5ad artifacts.
collection = ln.Collection.get(name="cellxgene-census", version="2024-07-01")
collection
Show code cell output
Collection(uid='dMyEX3NTfKOEYXyMKDD7', version='2024-07-01', is_latest=True, name='cellxgene-census', hash='nI8Ag-HANeOpZOz-8CSn', visibility=1, created_by_id=1, transform_id=22, run_id=27, created_at=2024-07-16 12:14:38 UTC)
You can count all contained artifacts or get them as a dataframe.
collection.artifacts.count()
Show code cell output
812
collection.artifacts.df().head() # not tracking run & transform because read-only instance
Show code cell output
! no run & transform got linked, call `ln.track()` & re-run
! run input wasn't tracked, call `ln.track()` and re-run
| uid | version | is_latest | description | key | suffix | type | size | hash | n_objects | n_observations | _hash_type | _accessor | visibility | _key_is_virtual | storage_id | transform_id | run_id | created_at | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | ||||||||||||||||||||
| 3042 | GcVBvpW5MYlrsH1izOjN | 2024-07-01 | True | All cells | cell-census/2024-07-01/h5ads/3dc61ca1-ce40-46b... | .h5ad | dataset | 947738392 | NDhyYVxRpOG6UiEkDZKswg | None | 71752 | md5-n | AnnData | 1 | False | 2 | 22 | 27 | 2024-07-12 12:34:09.211282+00:00 | 1 |
| 3587 | 1AeEHLQzGyRZL5nwpffu | 2024-07-01 | True | wilms | cell-census/2024-07-01/h5ads/ea01c125-67a7-4bd... | .h5ad | dataset | 75413467 | TNsJMqhUOekqUh4qtxvccA | None | 4636 | md5-n | AnnData | 1 | False | 2 | 22 | 27 | 2024-07-12 12:34:10.235938+00:00 | 1 |
| 2850 | vEw6vGy47Zi0Qj6TG6l7 | 2024-07-01 | True | Tabula Sapiens - Skin | cell-census/2024-07-01/h5ads/0041b9c3-6a49-4bf... | .h5ad | dataset | 199210144 | sV0vZMpxZsTXIb6qqCg8ng | None | 9424 | md5-n | AnnData | 1 | False | 2 | 22 | 27 | 2024-07-12 12:34:08.791875+00:00 | 1 |
| 3230 | tggrprv4cllqGOrH8RlL | 2024-07-01 | True | Dissection: Amygdaloid complex (AMY) - Basolat... | cell-census/2024-07-01/h5ads/7d3ab174-e433-40f... | .h5ad | dataset | 330480233 | eS_gAyJD_P0oLd6IHEsPJQ | None | 28984 | md5-n | AnnData | 1 | False | 2 | 22 | 27 | 2024-07-12 12:34:09.564663+00:00 | 1 |
| 3309 | RCzyhZz9tfi6YI4F7mxb | 2024-07-01 | True | Single cell RNA sequencing of follicular lymphoma | cell-census/2024-07-01/h5ads/99950e99-2758-41d... | .h5ad | dataset | 749041844 | FaUU0Z0Uk6w2oewwJq8zZg | None | 137147 | md5-n | AnnData | 1 | False | 2 | 22 | 27 | 2024-07-12 12:34:09.708223+00:00 | 1 |
You can query across artifacts by arbitrary metadata combinations, for instance:
query = collection.artifacts.filter(
organisms=organisms.human,
cell_types__in=[cell_types.dendritic_cell, cell_types.neutrophil],
tissues=tissues.kidney,
ulabels=suspension_types.cell,
experimental_factors=experimental_factors.ln_10x_3_v2,
)
query = query.order_by("size") # order by size
query.df().head() # convert to DataFrame
Show code cell output
| uid | version | is_latest | description | key | suffix | type | size | hash | n_objects | n_observations | _hash_type | _accessor | visibility | _key_is_virtual | storage_id | transform_id | run_id | created_at | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | ||||||||||||||||||||
| 2961 | WwmBIhBNLTlRcSoBDt76 | 2024-07-01 | True | Mature kidney dataset: immune | cell-census/2024-07-01/h5ads/20d87640-4be8-487... | .h5ad | dataset | 45158726 | GCMHkdQSTeXxRVF7gMZFIA | None | 7803 | md5-n | AnnData | 1 | False | 2 | 22 | 27 | 2024-07-12 12:34:09.039540+00:00 | 1 |
| 2961 | WwmBIhBNLTlRcSoBDt76 | 2024-07-01 | True | Mature kidney dataset: immune | cell-census/2024-07-01/h5ads/20d87640-4be8-487... | .h5ad | dataset | 45158726 | GCMHkdQSTeXxRVF7gMZFIA | None | 7803 | md5-n | AnnData | 1 | False | 2 | 22 | 27 | 2024-07-12 12:34:09.039540+00:00 | 1 |
| 3000 | gHlQ5Muwu3G9pvFCx3x8 | 2024-07-01 | True | Fetal kidney dataset: immune | cell-census/2024-07-01/h5ads/2d31c0ca-0233-41c... | .h5ad | dataset | 64546349 | 2qy8uy-65Sd_XcBU-nrPgA | None | 6847 | md5-n | AnnData | 1 | False | 2 | 22 | 27 | 2024-07-12 12:34:09.128217+00:00 | 1 |
| 3324 | P4Oai3OLGAzRwoicHfLM | 2024-07-01 | True | Mature kidney dataset: full | cell-census/2024-07-01/h5ads/9ea768a2-87ab-46b... | .h5ad | dataset | 194047623 | aZVpGZwAfMCziff_5ow2bg | None | 40268 | md5-n | AnnData | 1 | False | 2 | 22 | 27 | 2024-07-12 12:34:09.732579+00:00 | 1 |
| 3324 | P4Oai3OLGAzRwoicHfLM | 2024-07-01 | True | Mature kidney dataset: full | cell-census/2024-07-01/h5ads/9ea768a2-87ab-46b... | .h5ad | dataset | 194047623 | aZVpGZwAfMCziff_5ow2bg | None | 40268 | md5-n | AnnData | 1 | False | 2 | 22 | 27 | 2024-07-12 12:34:09.732579+00:00 | 1 |
Slice a concatenated array¶
Let us now use the concatenated version of the Census collection: a tiledbsoma array that concatenates all AnnData arrays present in the collection we just explored. Slicing tiledbsoma works similar to slicing DataFrame or AnnData.
value_filter = (
f'{features.tissue} == "{tissues.brain.name}" and {features.cell_type} in'
f' ["{cell_types.microglial_cell.name}", "{cell_types.neuron.name}"] and'
f' {features.suspension_type} == "{suspension_types.cell.name}" and {features.assay} =='
f' "{assays.ln_10x_3_v3}"'
)
value_filter
'tissue == "brain" and cell_type in ["microglial cell", "neuron"] and suspension_type == "cell" and assay == "10x 3\' v3"'
Query for the tiledbsoma array store that contains all concatenated expression data. It’s a new dataset produced by concatenating all AnnData-like artifacts in the Census collection.
census_artifact = ln.Artifact.get(description="Census 2024-07-01")
Run the slicing operation.
human = "homo_sapiens" # subset to human data
# open the array store for queries
with census_artifact.open() as store:
# read SOMADataFrame as a slice
cell_metadata = store["census_data"][human].obs.read(value_filter=value_filter)
# concatenate results to pyarrow.Table
cell_metadata = cell_metadata.concat()
# convert to pandas.DataFrame
cell_metadata = cell_metadata.to_pandas()
cell_metadata.head()
Show code cell output
! run input wasn't tracked, call `ln.track()` and re-run
| soma_joinid | dataset_id | assay | assay_ontology_term_id | cell_type | cell_type_ontology_term_id | development_stage | development_stage_ontology_term_id | disease | disease_ontology_term_id | ... | tissue | tissue_ontology_term_id | tissue_type | tissue_general | tissue_general_ontology_term_id | raw_sum | nnz | raw_mean_nnz | raw_variance_nnz | n_measured_vars | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 48182177 | c888b684-6c51-431f-972a-6c963044cef0 | 10x 3' v3 | EFO:0009922 | microglial cell | CL:0000129 | 68-year-old human stage | HsapDv:0000162 | glioblastoma | MONDO:0018177 | ... | brain | UBERON:0000955 | tissue | brain | UBERON:0000955 | 15204.0 | 3959 | 3.840364 | 209.374207 | 27229 |
| 1 | 48182178 | c888b684-6c51-431f-972a-6c963044cef0 | 10x 3' v3 | EFO:0009922 | microglial cell | CL:0000129 | 68-year-old human stage | HsapDv:0000162 | glioblastoma | MONDO:0018177 | ... | brain | UBERON:0000955 | tissue | brain | UBERON:0000955 | 39230.0 | 5885 | 6.666100 | 875.502870 | 27229 |
| 2 | 48182185 | c888b684-6c51-431f-972a-6c963044cef0 | 10x 3' v3 | EFO:0009922 | microglial cell | CL:0000129 | 68-year-old human stage | HsapDv:0000162 | glioblastoma | MONDO:0018177 | ... | brain | UBERON:0000955 | tissue | brain | UBERON:0000955 | 9576.0 | 2738 | 3.497443 | 121.333753 | 27229 |
| 3 | 48182187 | c888b684-6c51-431f-972a-6c963044cef0 | 10x 3' v3 | EFO:0009922 | microglial cell | CL:0000129 | 68-year-old human stage | HsapDv:0000162 | glioblastoma | MONDO:0018177 | ... | brain | UBERON:0000955 | tissue | brain | UBERON:0000955 | 19374.0 | 4096 | 4.729980 | 464.331956 | 27229 |
| 4 | 48182188 | c888b684-6c51-431f-972a-6c963044cef0 | 10x 3' v3 | EFO:0009922 | microglial cell | CL:0000129 | 68-year-old human stage | HsapDv:0000162 | glioblastoma | MONDO:0018177 | ... | brain | UBERON:0000955 | tissue | brain | UBERON:0000955 | 8466.0 | 2477 | 3.417844 | 162.555950 | 27229 |
5 rows × 28 columns
Create an AnnData object.
from tiledbsoma import AxisQuery
with census_artifact.open() as store:
experiment = store["census_data"][human]
adata = experiment.axis_query(
"RNA",
obs_query=AxisQuery(value_filter=value_filter)
).to_anndata(
X_name="raw",
column_names={
"obs": [
features.assay,
features.cell_type,
features.tissue,
features.disease,
features.suspension_type,
]
}
)
adata.var = adata.var.set_index("feature_id")
adata
! run input wasn't tracked, call `ln.track()` and re-run
AnnData object with n_obs × n_vars = 66418 × 60530
obs: 'assay', 'cell_type', 'tissue', 'disease', 'suspension_type'
var: 'soma_joinid', 'feature_name', 'feature_length', 'nnz', 'n_measured_obs'
Train ML models¶
You can either directly train ML models on very large collections of AnnData-like artifacts or on a single concatenated tiledbsoma-like artifact. For pros & cons of these approaches, see this blog post.
On a collection of arrays¶
mapped() caches AnnData objects on disk and creates a map-style dataset that performs a virtual join of the features of the underlying AnnData objects.
from torch.utils.data import DataLoader
census_collection = ln.Collection.get(name="cellxgene-census", version="2024-07-01")
dataset = census_collection.mapped(obs_keys=[features.cell_type], join="outer")
dataloader = DataLoader(dataset, batch_size=128, shuffle=True)
for batch in dataloader:
pass
dataset.close()
For more background, see Train a machine learning model on a collection.
On a concatenated array¶
You can create streaming PyTorch dataloaders from tiledbsoma stores using cellxgene_census package.
import cellxgene_census.experimental.ml as census_ml
store = census_artifact.open()
experiment = store["census_data"][human]
experiment_datapipe = census_ml.ExperimentDataPipe(
experiment,
measurement_name="RNA",
X_name="raw",
obs_query=AxisQuery(value_filter=value_filter),
obs_column_names=[features.cell_type],
batch_size=128,
shuffle=True,
soma_chunk_size=10000,
)
experiment_dataloader = census_ml.experiment_dataloader(experiment_datapipe)
for batch in experiment_dataloader:
pass
store.close()
For more background see this guide.